
Chromatographie Liquide Haute Performance (HPLC)
La chromatographie liquide haute performance (HPLC) ou simplement La chromatographie en phase liquide (CPL) ou liquid chromatography (LC) est une technique séparative utilisée en analyse quantitative, qualitative et principalement employée dans le domaine de la chimie analytique comme outil scientifique majeur mais aussi dans des domaines variés tels que la Toxicologie et la Biochimie.
Le champ d’application de l’HPLC recouvre une grande partie du domaine de la chromatographie en phase gazeuse auquel s’ajoute celui de l’analyse des composés thermosensibles ou de masses moléculaires à la fois très grandes et même polaires. Son succès est dû à la possibilité d’agir de manière très précise sur la sélectivité entre les composés par le choix de la colonne et de la composition de l’éluant, c’est-à-dire en exploitant les interactions soluté/phase mobile/phase stationnaire. L’efficacité des colonnes est moindre qu’en CPG, mais l’utilisation de phases chirales ou des nouvelles phases stationnaires opérant suivant plusieurs modes, les techniques par appariement d’ions ainsi que d’interaction hydrophobe accroissent encore plus les possibilités de la CLHP. Enfin la miniaturisation de la technique (nanochromatographie) a facilité son association avec la spectrométrie de masse.
Sommaire
Histoire
La chromatographie classique sur colonne a évolué au fil des ans, avec des innovations introduites à intervalles d’environ dix ans en matière de séparation chromatographique. Les techniques basée sur la chromatographie offraient des améliorations majeures en termes de vitesse, de puissance de résolution, de détection, de quantification, de commodité et d’applicabilité à de nouveaux types d’échantillons. La plus notable de ces modifications était la chromatographie liquide haute performance (HPLC). Les techniques modernes de CLHP sont devenues disponibles en 1969; Cependant, ils n’étaient pas largement acceptés dans l’industrie pharmaceutique jusqu’à plusieurs années plus tard. Une fois que les systèmes HPLC capables d’une analyse quantitative sont devenus disponibles dans le commerce, leur utilité dans l’analyse pharmaceutique a été pleinement appréciée.
Dans les années 1990, la HPLC avait entamé une croissance explosive qui en se faisait la méthode analytique la plus populaire jugée en fonction des ventes d’instruments et de l’importance scientifique. Sa popularité actuelle résulte de sa séparation pratique d’une large gamme de types d’échantillons, d’une puissance de résolution exceptionnelle, de la vitesse et des niveaux de détection nanomolaires. Il est actuellement utilisé dans la recherche pharmaceutique et le développement:
- Pour purifier les produits synthétiques ou naturels.
- Caractériser les métabolites.
- Pour doser les ingrédients actifs, les impuretés, les produits de dégradation et dans les essais de dissolution.
- Dans les études pharmacodynamiques et pharmacocinétiques.
Les améliorations apportées à la CLHP au cours des dernières années comprennent:
- Changements dans le matériau d’emballage, tels que la taille des particules plus petites, les nouveaux matériaux d’emballage et de colonne.
- Séparation à haute vitesse.
- Micro-HPLC, automatisation et optimisation assistée par ordinateur.
Amélioration des méthodes de détection, y compris les systèmes de détection dits « coupés ».
Origine de la CLHP (HPLC)
La chromatographie liquide haute performance, souvent désignée par son abréviation CLHP – HPLC en anglais –, constitue une technique analytique très générale d’emploi. Elle dérive de la forme la plus ancienne de la chromatographie liquide sur colonne dont les performances, en termes de sélectivité et de résolution, se sont trouvées grandement améliorées par la miniaturisation et l’utilisation de phases stationnaires très élaborées.
Ces phases, constituées de la réunion de micro-particules sphériques dont le diamètre est compris entre 2 et 5 micromètres ou de matériaux monolithiques poreux conduisent à une perte de charge importante dans la colonne. Il faut donc exercer sur la phase mobile une forte pression pour obtenir un débit convenable. Pour marquer cette particularité de la technique, la lettre P du sigle CLHP a pendant longtemps correspondu au mot pression.
La migration forcée d’une phase liquide au contact d’une phase stationnaire se retrouve dans plusieurs techniques chromatographiques. La particularité de la CLHP est de faire intervenir des mécanismes d’échange soluté/phase mobile/phase stationnaire basés sur les coefficients d’adsorption ou de partage.
Conception générale d’un appareil de HPLC (CLHP)
Une installation de CLHP (HPLC) comporte divers modules spécialisés, qui se présentent dans des boîtiers distincts ou intégrés dans un même châssis pour des raisons de moindre encombrement (fig. 1.1).
Ces modules sont reliés entre eux par l’intermédiaire de canalisations de très faible diamètre interne (0,1 mm) pour assurer la circulation de la phase mobile. Elles peuvent être en acier inoxydable ou en PEEK ® (ou polyether-etherketone), un polymère souple et coloré qui résiste aux solvants usuels, même sous des pressions élevées (350 bars).
L’écoulement des faibles débits obéit à la loi de Poiseuille. La vitesse de la phase mobile est maximum au centre des canalisations et nulle au contact des parois. Une dispersion des composés se produit donc inévitablement. Pour améliorer les séparations on fait donc en sorte que le volume de phase mobile hors-colonne soit le plus réduit possible (10 % du volume mort de la colonne).

Figure. 1 Schéma d’une installation de CLHP avec double détection. Un exemple de réalisation de type modulaire.
L’utilisateur compose son installation en fonction des applications prévues. La présentation en colonne des différents modules est commune à de très nombreux modèles concurrents. Ici le chromatographe modèle HP 1100 (reproduit avec l’autorisation de la société Agilent Technologies), comporte un injecteur automatique permettant un fonctionnement en continu et une colonne thermostatée pour améliorer la reproductibilité des séparations. Les composés élués, après passage par le détecteur UV sont identifiés avec encore plus de certitude au moyen d’un spectromètre de masse, situé à droite de l’image.

Figure 2. Appareil d’HPLC – Conception générale
Pompe et gradients d’élution
Pompes pour éluants
Toute installation de CLHP comporte au moins une pompe pour forcer le passage de la phase mobile à travers la colonne dont le remplissage est très compact. Il en résulte une pression importante au niveau de l’injecteur. Celle-ci peut atteindre 20 000 kPa (200 bars) selon le débit imposé à la phase mobile ou sa viscosité ainsi que selon la nature de la phase stationnaire.
On utilise des pompes conçues pour maintenir un débit non pulsé et stable, même si la composition de la phase mobile varie. Ces pompes débit-métriques comportent généralement deux pistons : en série (fig. 2).
C’est la partie qui sert à stocker l’éluant et à l’injecter sous pression dans la colonne. Elle est composée de :
- Deux pistons alternatifs fonctionnant en opposition pour éviter les interruptions de débit dues au remplissage du cylindre. Le déplacement des pistons est contrôlé par un moteur pas à pas associé à une came de forme particulière.
- Réservoirs de phase mobile
- Électrovannes
- Amortisseur de pulsations
- Système de purge et d’amorçage
- Capteur de pression
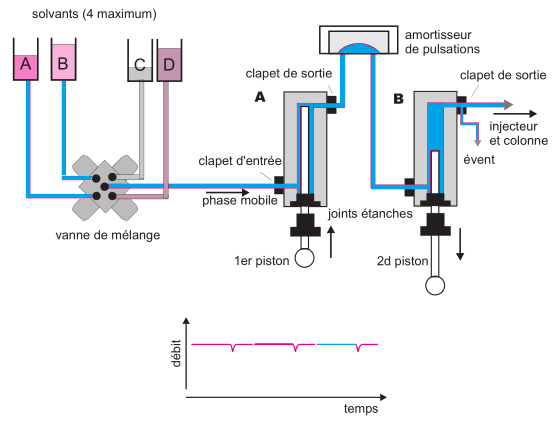
Figure 3. Principe de fonctionnement d’une pompe à deux têtes en série.
De manière simplifiée et en ignorant les corrections de compressibilité des solvants, on peut décrire ainsi le cycle de fonctionnement. Partant de l’instant où le clapet de sortie du cylindre A vient de se fermer et le clapet d’entrée vient de s’ouvrir, le piston de A recule pour remplir la chambre A. Pendant ce temps le cylindre B est ouvert et le piston de B avance pour chasser la phase mobile vers la colonne. Le volume déplacé par le piston de B est deux fois plus petit que le volume aspiré par le piston de A. Arrivé au fond de sa course, le clapet d’entrée de A se ferme et le clapet de sortie s’ouvre.
Le piston de A avance et chasse le contenu du cylindre A. Ce volume pour moitié est expulsé vers la colonne, l’autre moitié servant à remplir le cylindre B dans sa phase de recul. Entre les deux cylindres est placé un amortisseur de pulsations (dessin réalisé à partir d’un document de la société Agilent-Technologies). (En bas), Variations de débit
d’une pompe en fonction des cycles.
Injecteur
L’injection d’un volume précis de l’échantillon en tête de colonne doit se faire en un temps bref afin de perturber le moins possible le régime de circulation de la phase mobile qui doit être stable de la colonne au détecteur. On utilise pour ce faire, une vanne haute pression à plusieurs voies, manuelle ou motorisée dans le cas des injecteurs automatiques, placée juste avant la colonne (fig. 3). Il s’agit d’une pièce de précision qui doit résister à des pressions
pouvant dépasser 30 000 kPa. Elle fonctionne en deux temps :
➤ Dans la position chargement, où seule la communication entre pompe et colonne est assurée (fig. 3), l’échantillon est introduit à pression atmosphérique à l’aide d’une seringue dans un petit volume tubulaire appelé boucle. Celle-ci, dont il existe tout un choix de volumes, est soit extérieure, soit intégrée dans le corps de la vanne.
➤ Dans la position injection, l’échantillon est inséré dans le flux de phase mobile par rotation de 60◦ d’un levier qui permet d’inverser le sens de circulation dans la boucle. Une bonne reproductibilité des volumes n’est atteinte que si la boucle a été totalement remplie par l’échantillon. Le volume prélevé avec la seringue est donc toujours largement
supérieur à celui de la boucle.
Des tubes en acier inoxydable, en Teflon, en PEEK ou en silice fondue permettent de relier la ou les pompes à l’injecteur chromatographique. Il y a plusieurs types d’injecteurs :
- Boucle d’injection : permet la répétabilité du volume d’injection
- Injecteur seringue
- Extraction sur phase solide en ligne

Figure 4. Vanne d’injection pour CLHP et boucles assorties. Vanne vue de l’arrière (vanne à 6 entrées/sorties avec une boucle raccordée) et assortiment de boucles de différents volumes.

Figure 5. Injection avec une boucle. a) Remplissage de la boucle. Dans cette étape, la seringue est introduite à la position n ◦ 4; b) injection dans la colonne (noter la nouvelle position de la manette). Vanne modèle 7125. Les vannes sont motorisables.
Colonnes
La colonne se présente comme un tube, le plus souvent en acier, dont la longueur et le diamètre présentent des différences selon les modèles. Les colonnes « standard » dont le diamètre interne (DI) est d’environ 4,5 mm et la longueur de 10 cm (fig. 5), sont de plus en plus supplantées par des colonnes de plus faibles diamètres, baptisées narrow-bore (DI 2-4 mm), micro-bore (DI 1-2 mm), capillaires remplies (DI 0,1-1 mm). Ces modèles sont apparus suite à l’évolution des phases stationnaires customisées et pour simplifier les problèmes de couplage avec la spectrométrie de masse.
La colonne est souvent précédée d’une précolonne, dite colonne de garde, courte (0,4 à 1 cm), remplie de la même phase stationnaire, ce qui sert à retenir certaines impuretés (fig. 5). On augmente ainsi la durée de vie de la colonne principale en préservant ses performances.

Figure 6. Colonne standard et précolonne de CLHP. Aspects extérieurs éclatés et assemblés d’une colonne ZORBAX ® . La phase stationnaire est maintenue par deux disques poreux situés à ses extrémités. La surface interne du tube est rendue inerte par un traitement de passivation, ou par un chemisage de verre ou de polymère PEEK ® ). La précolonne, périodiquement changée, évite le colmatage de la colonne. Il n’empêche qu’il est conseillé de faire passer les échantillons avant analyse à travers un filtre de porosité inférieure à 0,5 mm (reproduit avec l’autorisation de la
société RTI).
Effet de Modification de la température
Les écarts de température modifiant les temps de rétention, les appareils actuels permettent de thermostater la colonne et l’éluant, à la fois pour assurer la répétitivité des analyses et pour faire intervenir éventuellement la température comme paramètre de séparation (fig. 6).

Figure 7. Effet de la température de la colonne sur une séparation. Exemple relatif à trois essais effectués sur un même mélange et avec un même débit de phase mobile à des températures différentes (a) 25 ◦ C, (b) 35 ◦ C et (c) 45 ◦ C
Phases stationnaires
La recherche d’une bonne résolution chromatographique et par voie de conséquence d’une efficacité élevée, a conduit à la création de phases stationnaires de nature et de structures variées. Pour raccourcir les temps d’analyse, il faut tenter d’accélérer dans la colonne les transferts entre les phases mobile et fixe.
Le gel de silice, matière de base des phases actuelles
Parmi tous les matériaux qui ont été ou sont actuellement utilisés pour la confection des phases stationnaires, le gel de silice tient une place prépondérante. Ce matériau de base est un solide amorphe ayant pour formule de composition SiO2 (H2O)n (avec n très proche de 0). Il est tout à fait différent de la silice naturelle cristalline (SiO2 ) qui n’est qu’un précurseur très éloigné de sa préparation. Cette dernière fait appel à des procédés de polymérisation sol-gel d’un tétraalcoxysilane (ex. tétraéthoxy-silane) au sein d’un liquide, sous l’effet d’une hydrolyse catalysée (fig. 7.)
Le gel de silice n’a pas la structure ordonnée de la silice cristalline, mais il reste néan-moins bâti autour de l’agencement tétraédrique des quatre liaisons issues de l’atome de silicium. C’est un polymère inorganique réticulé. Il comporte des groupements silanols , Si–OH en nombre variable, qui ont résisté à la phase finale de déshydratation thermique.
Ces groupements sont responsables des propriétés catalytiques acides de ce matériau très polaire car Si–OH a un pK de 10, comparable à celui du phénol. Leur concentration peut être établie par RMN du 29Si ou par analyse centésimale du carbone pour les phases greffées.

Figure 8. Le gel de silice pour chromatographie. a) préparation de grains sphériques de gel de silice via un sol-gel. La dispersion, appelée sol, constituée de particules sphériques de quelques nm de diamètre, s’agglutine en présence d’un liant organique urée/formol jusqu’à atteindre la taille voulue (3-7 mm). Le traitement final consiste en une pyrolyse pour éliminer la matrice organique. b) représentation du réseau, correspondant à un maillage tridimensionnel, d’un gel de silice porteur de groupements silanols. c) image d’une particule sphérique de gel de silice issus d’un assemblage compact de sphères submicroniques.
Porosité
Le gel de silice comporte des pores de tailles différentes. Pour remplir la colonne d’une manière homogène, il est préparé sous forme, soit de microparticules sphériques, soit d’un monolithe poreux (fig.8 et 10).Il est nécessaire en effet d’éviter la formation de chemins préférentiels pour la phase mobile et par suite pour les composés qui y sont dissous.
➤ Les micro-sphères ont un diamètre constant dans une même colonne mais il en existe plusieurs types allant de 1 à 12 mm.
➤ Les monolithes, apparus plus récemment, sont ainsi nommés parce qu’il s’agit d’un gel de silice formé d’une seule pièce dans la colonne même. La reproductibilité des caractéristiques de ce second type de colonne est plus difficile à maîtriser.

Figure 9. Représentations imagées de quelques types de gels de silice (porosité et dimensions). 1- structure comportant des pores de diffusion répartis dans la totalité de la particule. 2- structure comportant des fissures de perfusion pour accélérer le processus de transfert. 3 et 4- Particules poreuse en surface avec un noyau non poreux. 5- détail de structure d’un remplissage du type monolithique.
Les silices greffées
Bien qu’ayant une capacité d’adsorption élevée, le gel de silice décrit précédemment n’est plus utilisé tel quel en chromatographie analytique. Hydrophile par nature, ses caractéristiques évoluent au cours du temps, entraînant un manque de reproductibilité des séparations.
Pour diminuer sa polarité jugée excessive dans de nombreux cas on le rend essentiellement hydrophobe. Les modifications classiques mettent à profit la réactivité des fonctions silanols présentes en surface pour fixer des molécules organiques par des liaisons covalentes. Le gel de silice ainsi modifié devient assimilable à un liquide immobilisé, la séparation mettant en jeu les coefficients de partage et non plus les coefficients d’adsorption. Ces phases greffées, dont lapolarité peut être ajustée avec précision, sont à l’origine de la chromatographie de partage
à polarité de phase inversée, utilisée dans quasiment toutes les séparations.
Ces modifications de la surface du gel conduisent à deux types de phases:
Phases monomériques (10–15 µm d’épaisseur)
Elles sont obtenues en faisant réagir un monochlorosilane en présence d’une base sur les fonctions silanols de surface. On prépare ainsi les phases classiques RP–8 (groupement diméthyloctylsilane) et RP–18 (groupement diméthyloctadécylsilane, ou ODS). Une fraction des groupements Si-OH demeure cependant intacte. Elle peut être la cause d’interactions polaires gênantes. D’autres réactifs silylés tels le chlorotriméthylsilane (ClSiMe3) ou l’hexaméthyldisilazane (Me3SiNHSiMe3 ), conduisent à une réaction plus complète. Les quelques sites restants non
transformés, car inaccessibles au réactif, le sont également aux analytes.
Phases polymériques (25 µm ou plus en épaisseur).
On utilise cette fois un di- ou trichlo-rosilane en présence de vapeur d’eau qui provoque une polymérisation en solution du réactif avant dépôt et greffage sur la silice. On obtient ainsi une couche polymérique réticulée.
L’architecture finale du revêtement est difficile à se représenter. Mono- ou multicouche, sa représentation à l’échelle moléculaire relève plutôt de la spéculation.
Détecteur
Il existe plusieurs types de détecteurs :
- détecteurs spectroscopiques :
- par spectroscopie d’absorption : ultraviolet-visible ou infrarouge,
- par spectroscopie de fluorescence ;
- réfractométrie ;
- détecteurs électrochimiques (DEC) :
- ampérométriques,
- coulométriques,
- polarographiques,
- potentiométriques ;
- conductimétrie ;
- UV à barrette de diodes (DAD) ;
- évaporatif à diffusion de la lumière (DEDL) ;
- détection spectrale avec couplage :
- à la spectrométrie de masse (MS), exemples : chromatographie en phase gazeuse-spectrométrie de masse et chromatographie en phase liquide-spectrométrie de masse ;
- à la spectroscopie atomique.

Figure 10. Détecteur HPLC
Détecteurs à barrettes de photodiodes
Le détecteur à barrette de photodiodes (DAD) est un détecteur UV avancé. En fonction de la longueur d’onde, une lampe au tungstène et une lampe au deutérium sont utilisées comme sources lumineuses. Le faisceau de lumière polychromatique est focalisé sur une cellule d’écoulement (volume de 8 à 13 μL) et ensuite dispersé par un réseau holographique ou un prisme de quartz. La lumière spectrale atteint alors une puce qui contient 100 à 1000 diodes photosensibles disposées côte à côte. Chaque diode enregistre seulement une fraction bien définie de l’information et de cette manière toutes les longueurs d’onde sont mesurées en même temps. Il convient de noter que si la présence de plus de diodes dans un réseau augmente la résolution des spectres UV, elle diminue la sensibilité absolue car une moindre quantité de rayonnement est absorbée par chaque diode. La résolution en longueur d’onde des détecteurs les plus récents est de l’ordre de 1 nm par diode, avec une précision de longueur d’onde supérieure à ± 1 nm et une sensibilité inférieure à 10-4 unités d’absorption. Toutes les opérations du détecteur sont commandées par un ordinateur: correction des fluctuations de l’énergie de la lampe, collecte des signaux (Iλ) de toutes les diodes, stockage des données de la phase mobile (I0λ, mesurée au début du chromatogramme) et calcul de l’absorbance selon la loi de Beer-Lambert de Iλ à I0λ. Le nombre de spectres enregistrés par seconde peut être choisi entre 0,1 et 10; habituellement, un spectre / seconde est optimal en ce qui concerne la résolution chromatographique et le bruit. En fin de parcours, un spectrochromatogramme tridimensionnel (absorbance en fonction de la longueur d’onde et du temps) est stocké sur l’ordinateur et peut être évalué qualitativement et quantitativement. Une description détaillée de l’opération DAD est donnée dans Huber et George (1993).
La détection des barrettes de diodes offre plusieurs avantages. La connaissance des spectres des composés d’intérêt permet d’éliminer les pics d’interférence de sorte qu’une quantification précise des pics d’intérêt peut être obtenue malgré une résolution moins qu’optimale. La détection simultanée à deux longueurs d’onde permet de calculer un rapport d’absorbance. Si ce rapport n’est pas constant à travers un pic, le pic n’est pas pur, quel que soit son aspect. Un avantage supplémentaire de la détection par réseau de diodes est la soustraction d’une longueur d’onde de référence. Cela réduit la dérive de la ligne de base pendant l’élution du gradient. Les systèmes HPLC-DAD liés aux banques de spectres UV sont particulièrement utiles en toxicologie clinique et médico-légale pour le criblage de médicaments dans des échantillons biologiques et son utilisation dans ce contexte est décrite plus loin (Pragst et Herzler, communication personnelle) .Refractive index detectorThe RI le détecteur est un détecteur universel, en ce sens que les changements de RI (positifs ou négatifs) qui résultent de la présence d’un composé dans l’éluant sont enregistrés. Cependant, c’est aussi le détecteur le moins sensible (jusqu’à 100 fois moins sensible que la détection UV). Des détecteurs d’IR peuvent être utilisés pour des excipients tels que des sucres dans des produits pharmaceutiques. De nombreux facteurs influencent l’IR et doivent être contrôlés pendant la séparation, tels que la température, la composition de l’éluant et la pression.
Dégazeur
Le Dégazeur est un composant permet de retirer l’oxygène présent dans le(s) solvant(s) afin d’éviter d’endommager les échantillons ou la phase stationnaire. Deux types de dégazeurs sont utilisés en HPLC :
- Dégazeur à gaz inerte : On fait barboter un gaz inerte dans la PM (Phase Mobile) pour retirer le gaz dissous dans le liquide. L’helium est le gaz inerte le plus utilisé pour cette application.
- Dégazeur par vide : Cette méthode consiste à descendre en pression dans une enceinte où se trouve le solvant à l’aide d’une pompe à vide primaire, et ainsi séparer le gaz dissous dans le fluide. Elle est bien plus efficace, ne requiert plus de gaz inerte, et remplace de plus en plus l’ancienne technique dans le domaine de l’analyse HPLC. La pression dans l’enceinte est de l’ordre du millibar.
Source : Columns: Clarke’s Analysis of Drugs and Poisons 3rd Edition.